Thermodynamic properties of solid solutions determine their behaviour in contact with aqueous solutions. Deviations of their thermodynamics from thermodynamics of ideal solutions is the most important factor in the equilibria in systems of several solid solutions and liquid. Models of multi-component solid solutions convenient for reliable calculations must be flexible to be able to describe different types of behaviour of solid solutions but should involve as few as possible numerical parameters to be fitted on few experimental data. Cluster-variation theory of solid solutions with several different cation site types allows one to develop quite accurate description of thermodynamics and ordering in minerals. We present here description of ternary solid solutions with two different cation site types. The mathematical part of the theory will be published in detail in Ivanov (1996). (The same theory for binary solid solutions and its application to Fe-Mg orthopyroxenes; see Ivanov, 1991). In our approach we take pairs of atoms as the maximal clusters. The latter limitation allows us to describe multi-component solid solutions using only parameters which characterise energies of atoms and their pairs in binary components of the multi-component solid solution. In the binary solid solutions with two different kinds of sites the number of these energetic parameters does not exceed 4. The model allows to describe most of important types of behaviour of solid solutions (short- and long-range ordering and disordering, asymmetry, incomplete mixing and separation into two phases, and some other phenomena). For n-component solid solutions 3n(n-1)/2 parameters are needed for orthopyroxene lattice and 2n(n-1) for olivine. In Fig. 1 we present the 3-D plot of Gibbs excess mixing energy for a solid solution with orthopyroxene crystal structure where for AB pair the real energies of Fe-Mg interaction are taken (Ivanov, 1991). As an example of the application of the theory to real solid solutions we present thermodynamics of Ni-Mg olivines. The set of cation pairs with energies which can affect the thermodynamic properties of the crystal is determined through the analysis of the inter-cation distances. This determines the number of energetic parameters (4), needed to describe ordering and thermodynamics of Ni-Mg olivines. The available experimental X-ray data on cation ordering in Ni-Mg olivines allow us to determine three of these parameters. The whole set of parameters is determined by a fitting procedure on the base of X-ray data combined with available experimental data on olivine-oxide equilibrium. The values of parameters are W11=324, W12=555, W22=853 (the short-range parameters), and W1Ni = -6609 cal/mol (the difference of the site-preference energies). This results in a small positive deviation of the solid solution from the ideal mixing (Fig. 2).
Ivanov, M.V., Geochemistry International 28(6), 117-126 (1991).
Ivanov, M.V., Crystallography Reports, (1996, in press).
Fig. 1.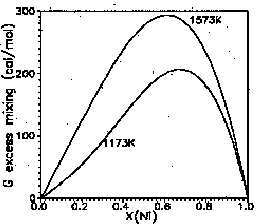
Fig. 2.