In many areas of geochemical modelling as well as in studies into the partitioning of elements, the family of the rare earth elements (REE) play an important part as indicators and tracers of processes. Their analysis is often constrained by restricted access to relatively expensive techniques, the larger sample size often required as well as by the interference of other elements. A similar problem is encountered with elements such as Be which are out of the range of most common analytical techniques or too low in concentration.
At the beginning of this decade a new analytical technique was introduced, capillary ion analysis (CIA) or capillary electrophoresis, as a fast and comparatively inexpensive technique. The instrumentation available now is space saving and relatively easy to operate. By its nature it is a chemically interactive, low cost analytical technique for cationic and anionic species. The separation is based on the different electrophoretic mobility of the hydrated or complexed species, or their charge/mass ratio, in an electrolyte moving through a fused silica capillary in an electric field (about 25 kV potential). The detection is accomplished at the end of the capillary by indirect UV detection. CIA allows two different modes of analyte introduction into the capillary, i) by hydrostatic pressure when the analyte containing vial is raised relative to the receiving vial (typical injection of about 100 nl) and, ii) by electromigration, when a smaller voltage (about 4-5kV) is applied between the sample vial and the receiving container. The latter mode is unique to CIA and not only allows the restricted intake of only one ionic species (cations or anions) but also causes an enrichment of the species at the tip of the capillary by a factor of 50 to 100 under normal conditions of analysis. This injection mode obviously provides an elegant method to lower the detection limits for most species. Dependent on the mass of the hydrated species detection limits in the lower ppb region and, in some cases in the ppt region can be achieved.
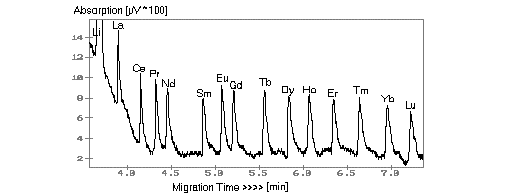
Since the first detection and separation of REE by CIA in the presence of Group I and II elements and transition metals in 1991, many improvements have been made to electrolyte compositions and improved base line separations. Results are reported here using a pyridine electrolyte with HIBA as complexing agent. The detection limit, using electromigration injection, was found to be between 2 and 5 ppb for the REE. It is apparent from Fig. 1 that the response of the REE to the separation is almost uniform because of their chemical behaviour. Using hydrostatic and electromigrative injections in sequential analyses a range of 3 to 4 magnitudes in composition can be accommodated without additional dilution or change of the analyte.
This analytical approach has been applied to the REE analysis of the so-called 'fly-speck'carbonaceous nodules of Witwatersrand conglomerates. The nodules are up to 2 mm in diameter, highly enriched in Th, U and the REE, and a conspicuous feature of many conglomerates. The low sample requirement for a CIA analysis, in this case about 1 to 2 mg, allows the analysis of single grains and thus a more detailed investigation into the nature of these nodules. Their REE pattern is characterised by a pronounced enrichment of the HREE.
In addition to the analysis of REE the technique has also been applied to very small samples (<1 mg) of various minerals. Examples are inclusions of villeaumite (NaF) in a phonolite from Namibia and small crystal aggregates of bavenite (Be) from the pegmatitic part of a Cape granite.
Fig. 1: Separation of REE by CIA for a 5 ppm standard. Electrolyte: 10 mM pyridine + 5 mM HIBA (pH4.5), voltage: +25 kV, capillary: 60 cm x 75 µm, hydrostatic injection, UV detection at 254 nm.